
Overview … Phase II ERT Clinical Trial …
The “EMBOLDEN” Study
updated 5-Feb-2021
Takeda (formerly Shire) is in the midst of a Phase II Clinical Trial to expand its evaluation of the efficacy of their SHP611 Enzyme Replacement Therapy (ERT) for MLD. They call this study the “Embolden” study. The trial launched in April 2019 and completed enrollment of 42 patients in January 2021, 18 months before the target completion in the summer of 2022. The study lasts 210 weeks (4 years). Participants will complete the formal trial period starting mid 2023 and continuing through early 2024.
We expect some initial data reporting to occur before the formal end of the trial, but formal public reporting of full trial results will likely not be public until a year after the study completes, perhaps early in 2026. Patients will likely be offered continuing access to enzyme while the Embolden data is compiled, analyzed and reported.
Compared to the Phase I/II trial they have increased the dose, increased the frequency, will be opening sites around the world including for the first time in the US, and will be including symptomatic late-infantile patients.
THERAPEUTIC APPROACH
The therapy is an Enzyme Replacement Therapy (ERT). Much like a diabetic receives regular insulin injections for diabetes, this is a weekly infusion (injection) of an enzyme designed to substitute for the body’s missing ARSA enzyme. ERT has been successfully used for other lysosomal diseases for over a decade.
INFUSION
 The drug delivery will be weekly through a small port (soft rubber-like button) that is placed under the skin via a minor surgery, usually on the front/side of the patient. From the port, a catheter (tube)
The drug delivery will be weekly through a small port (soft rubber-like button) that is placed under the skin via a minor surgery, usually on the front/side of the patient. From the port, a catheter (tube)
is connected to the spinal column which is a pathway for the enzyme infusions to flow freely into the brain. The cerebral spinal fluid (CSF) is on the other side of the blood-brain barrier and freely circulates through the spine to the brain. This permanent port eliminates the need for a lumbar puncture with each weekly infusion.
The infusions only take a few minutes, involve a quick needle prick into the port, and a bit of time post-injection to monitor for any reactions. The dose is 150mg (per kg), a higher and more frequent dose than the prior trial. The trial lasts 105 weeks (2 years) with an expectation that benefiting participants will be able to continue on the therapy after the formal trial ends as has been the case with the past studies.
COST
The drug and all study-related testing will be paid for by the trial sponsor. Families should discuss the need for additional travel support, if any, at the time of assessment and enrollment.
WHAT IS GMFC-MLD?
Gross Motor Function Classification System or GMFCS is a 5 level clinical classification system that describes the gross motor function of people on the basis of self-initiated movement abilities. Particular emphasis on creating and maintaining the GMFCS scale levels rests on evaluating sitting, walking, and wheeled mobility. Distinctions between levels are based on functional abilities; the need for walkers, crutches, wheelchairs, or canes/walking sticks; and to a much lesser extent, the actual quality of movement.
The original version of the GMFCS was developed in 1997 cerebral palsy. The current GMFC-88 has been adapted to the unique characteristics of MLD and is referred to as GMFC-MLD.
The following three graphics may help you to preliminarily self-determine what GMFCS-MLD level your loved one might be. The 3rd graphic provides more detailed descriptions broken out by age group from birth to 18 years old.


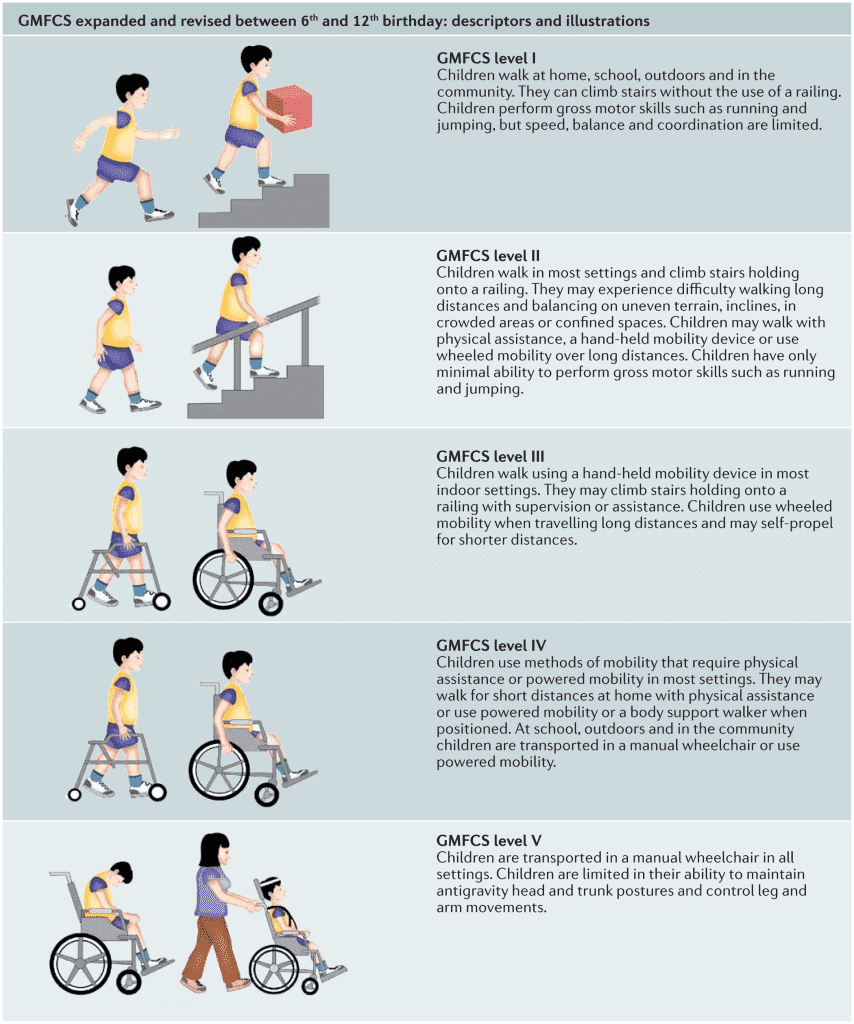
Click on the image below to enlarge it to show more details by age group from birth through age 18:

SUMMARY & ADDITIONAL DETAILS
Study Summary – Weekly intrathecal injections for 105 weeks via an intrathecal drug delivery device (IDDD) or port implanted in the patient’s abdomen.
What is a Clinical trial? – Clinical trials are research studies performed in people that are aimed at evaluating a medical, surgical, or behavioral intervention. They are the primary way that researchers find out if a new treatment, like a new drug or medical device (for example, a pacemaker) is safe and effective in people. Efficacy, while desired and measured, cannot be guaranteed.
Infusion Time – The weekly infusion is expected to take just a few minutes, but it is likely patients will be monitored for an hour or two after each infusion to watch for any allergic reactions – which are not likely but is part of understanding the drug.
Cohorts – The study will be performed with 35 patients divided into four cohorts at varying degrees of disease progression.
Matched Historical Controls – these are untreated patients that will be identified through a separate CHOP natural history study and matched to treated patients based on medical history and progression. This enables Takeda to compare the efficacy of therapy to the natural history of untreated disease.
Enzyme – Recombinant human arylsulfatase A, which is expected to work in the same way as normal human arylsulfatase using a method known as “recombinant DNA technology”: it is made by human cells that have received a gene (DNA) that makes them able to produce arylsulfatase A.
 Intrathecal Port – The spinal fluid naturally circulates between the spinal column and the brain 3.7 times per day and it is on the “other side” of the blood-brain barrier that is a typical barrier to an intravenously delivered enzyme.* This intrathecal therapy delivers the enzyme directly into the spinal fluid. In order to minimize the chances of infection or injury due to repeated spinal puncture, a small port (a little larger than a US dime in diameter) will be implanted in the abdomen just under the skin – usually on the patient’s side or front which is connected via a very small catheter to spine and into the CSF.* The Enzyme is injected through the skin into the port and transfers via the catheter (tubing ) to the spinal fluid. This port is being studied as an integral part of the Clinical Trial. Please note – this is a static bolus device, not a pump. The image is the new (December 2013) Sophysa mini Soph-A-Port®.
Intrathecal Port – The spinal fluid naturally circulates between the spinal column and the brain 3.7 times per day and it is on the “other side” of the blood-brain barrier that is a typical barrier to an intravenously delivered enzyme.* This intrathecal therapy delivers the enzyme directly into the spinal fluid. In order to minimize the chances of infection or injury due to repeated spinal puncture, a small port (a little larger than a US dime in diameter) will be implanted in the abdomen just under the skin – usually on the patient’s side or front which is connected via a very small catheter to spine and into the CSF.* The Enzyme is injected through the skin into the port and transfers via the catheter (tubing ) to the spinal fluid. This port is being studied as an integral part of the Clinical Trial. Please note – this is a static bolus device, not a pump. The image is the new (December 2013) Sophysa mini Soph-A-Port®.
Placing the port and catheter is a minor surgery and will require sedation. Please ask us about how to best sedate MLD patients for this surgery.
24 International & US Sites – United States (California, Florida, Illinois, Iowa, Minnesota, New York, Ohio, Pennsylvania, Utah) Belgium, Brazil, Canada, Denmark, France, Germany, Israel, Japan, Mexico, Netherlands, Spain, United Kingdom.
You may enroll and start therapy at one site and then transfer to another site closer to home as it opens.
==> Details for each site, including contact information, can be found here.
Local Infusion Sites – Enrollment and periodic assessments must be at a primary study site (see above), however, it is Takeda’s desire to enable the weekly infusions to occur locally, if appropriate local resources are available. These local resources will have to be evaluated, authorized, and trained on a case by case basis.
Continuation of Enzyme Access After the Primary Study Period – Takeda has indicated they will continue to supply of enzyme after the 106 week study period. Historically this has been done as part of a separate “continuation study” that all patients are automatically enrolled in.
Primary End Points – Decline or lack of decline in GMFC-MLD. Group A is expected to decline no more than 2 levels. Additional observations and tests will be for Expressive Language Function Classification in Metachromatic Leukodystrophy (ELFC-MLD), change from baseline in Cerebrospinal Fluid (CSF) sulfatides levels, etc.
Compassionate Use/Named Access – to the best of our knowledge no Compassionate Use or Named Access for SHP611 will be made for non-trial participants during the recruitment phase of this study.
Phase II Trial Time Frame – Takeda estimates the recruiting and two year study period will end in late 2022.
Development Timeline – This ERT has been in various stages of Clinical Trial since 2012.
Why is the Trial Targeted for Late Infantile MLD? Most MLD Clinical Trials target the late-infantile form of the disease because this form of the disease is the most rapidly progressing and hence it is easier and faster to show the efficacy of the enzyme therapy.
Access for Juveniles and Adults – It is Takeda’s desire, after drug approval, to make the ERT available to patients with all forms and stages of MLD where efficacy can be expected. Studying the ERT in late-infantiles is the fasted path to therapy approval by regulatory agencies (FDA, EMA, etc.) Please note, your treating doctor (not Takeda) may determine that your loved one is too advanced to see any benefit from the therapy.
ClinicalTrials.gov – Trial ID NCT03771898
Takeda Embolden Study Overview – Trial Overview provided by Takeda
Orphan Designation – SHP611 has Orphan designation by the EMA for Europe (EU/3/10/813).